Hyponatraemia
Last updated 20th March 2019 - Tom Heaton
Hyponatraemia can be a particularly challenging medical problem to deal with because of the interaction between sodium and water in both absolute and relative terms.
An overview of fluid and sodium physiology is therefore a useful starting point and can be found here: http://www.medicalphysiology.co.uk/fluid--electrolyte-overview.html
This is quite a nice introductory video on hyponatraemia: https://www.youtube.com/watch?v=dmM2K50bnKw
An overview of fluid and sodium physiology is therefore a useful starting point and can be found here: http://www.medicalphysiology.co.uk/fluid--electrolyte-overview.html
This is quite a nice introductory video on hyponatraemia: https://www.youtube.com/watch?v=dmM2K50bnKw
Definition
Hyponatraemia is a serum sodium concentration below the lower range of normal (this will vary on the laboratory/definition but is often defined as 135 mmol/L).
Acute hyponatraemia is when it has developed under 48 hours.
Chronic hyponatraemia refers to an onset time of over 48 hours.
The magnitude of the disturbance may also be arbritraily defined:
Acute hyponatraemia is when it has developed under 48 hours.
Chronic hyponatraemia refers to an onset time of over 48 hours.
The magnitude of the disturbance may also be arbritraily defined:
- <120 mmol/L - severe
- 120-130 mmol/L - moderate
- >130 mmol/L - mild
Pathophysiology
Sodium (Na+)is the most important extracellular cation.
As such, it plays a central role in the osmotic state of the body and is intrinsically linked with the water throughout the body.
It is therefore difficult to unpick the pathophysiology of sodium disturbances with understanding the normal physiology of sodium and water homeostasis.
The key components of this homeostasis are:
It is usually due to a failure to excrete water normally.
As the sodium is the main determinant of extracellular osmolality, hyponatraemia usually also represents a hypoosmolar state.
There are rarer cases where the osmolality may be normal or high due to the (usually pathological) increase in another osmotic molecule e.g. glucose.
As such, it plays a central role in the osmotic state of the body and is intrinsically linked with the water throughout the body.
It is therefore difficult to unpick the pathophysiology of sodium disturbances with understanding the normal physiology of sodium and water homeostasis.
The key components of this homeostasis are:
- Oral intake/thirst mechanisms
- Antidiuretic hormone
- Renin-angiotensin axis
- Renal handling of Na+
It is usually due to a failure to excrete water normally.
As the sodium is the main determinant of extracellular osmolality, hyponatraemia usually also represents a hypoosmolar state.
There are rarer cases where the osmolality may be normal or high due to the (usually pathological) increase in another osmotic molecule e.g. glucose.
Aetiology
There are a very wide range of potential causes.
They are probably best remembered by dividing them into the categories of overall volume status.
This gives:
Factitious
Here the ‘hyponatraemia’ is actually arising due to the interference of other osmotically significant substances.
Hypovolaemic
Euvolaemic
Hypervolaemic
The features of a few of the important causes, as well as the general features of these groups, are discussed in more detail below.
They are probably best remembered by dividing them into the categories of overall volume status.
This gives:
- Factitious
- Hypovolaemic
- Euvolaemic
- Hypervolaemic
Factitious
Here the ‘hyponatraemia’ is actually arising due to the interference of other osmotically significant substances.
- Pseudohyponatraemia - elevated proteins/lipids
- Hyperglycaemia
- Mannitol
- Toxic alcohols
- Post TURP - glycine
Hypovolaemic
- Vomiting
- Diarrhoea
- Skin losses - burns, excess sweating
- Diuretics
- Renal failure
- Cerebral salt wasting
- Adrenocorticoid deficiency
Euvolaemic
- SIADH
- Hypothyroidism
- Acute/chronic water load
- Tea & Toast/Beer diet
- Glucocorticoid deficiency
Hypervolaemic
- CCF
- Hepatic failure
- Renal failure
- Nephrotic syndrome
- Primary polydipsia
The features of a few of the important causes, as well as the general features of these groups, are discussed in more detail below.
Presentation
The symptoms of hyponatraemia will vary depending on the cause.
Two key factors that will impact upon this are:
A rapid drop can result in more cellular dysfunction due to fluid shifts, particularly cerebrally, and therefore be more symptomatic.
Symptoms may include:
Signs
The history of the presentation will be very important to aiding the overall diagnosis.
Although this may be quite apparent, domains of questioning that can be useful include:
Two key factors that will impact upon this are:
- Magnitude of Na+ fall
- Speed of Na+ fall
A rapid drop can result in more cellular dysfunction due to fluid shifts, particularly cerebrally, and therefore be more symptomatic.
Symptoms may include:
- CNS
- Lethargy
- Headache
- Confusion
- Personality change
- Muscle cramps
- Weakness
- Decreased conscious level
- Seizures
- GI
- Anorexia
- Nausea/vomiting
Signs
- CNS
- Confusion
- Cognitive impairment
- Seizures
- Herniation syndrome (severe acute hyponatraemia)
- CVS
- Hypovolaemic changes
- Dry mucous membrane
- Reduced skin turgor
- Hypovolemic changes
- Tissue oedema
- Raised JVP
- Ascites
- Pulmonary oedema
- Hypovolaemic changes
The history of the presentation will be very important to aiding the overall diagnosis.
Although this may be quite apparent, domains of questioning that can be useful include:
- History of electrolyte losses e.g. vomiting, diarrhoea
- Low intake states e.g. malnutrition, alcohol misuse
- Medication history
- History consistent with malignancy
- Recent surgery
Assessment
This is where the management of hyponatraemia can become challenging.
A number of investigations can be performed, but these need integrating with the history and clinical history to form an appropriate diagnosis or list of differentials.
Some investigations will be to specifically investigate possible causes e.g. TFTs.
A number of investigations can be performed, but these need integrating with the history and clinical history to form an appropriate diagnosis or list of differentials.
Some investigations will be to specifically investigate possible causes e.g. TFTs.
Investigations
Bloods:
ECG - hyponatraemia may cause non ischaemic ST elevation
CXR - this may provide evidence of pulmonary oedema or a lung malignancy.
- U&Es
- FBC
- LFTs
- Bone profile
- TFTs
- Cortisol - random or synacthen test
- Electrolytes (spot vs collection)
- Osmolarity
ECG - hyponatraemia may cause non ischaemic ST elevation
CXR - this may provide evidence of pulmonary oedema or a lung malignancy.
Analysis
Combining the clinical presentation with laboratory results can then allow an appropriate categorisation of likely causes.
This is often usefully demonstrated with a flow chart.
Essentially, this is guiding you through the key steps of the pathophysiology to help with understanding the underlying problem.
This is often usefully demonstrated with a flow chart.
Essentially, this is guiding you through the key steps of the pathophysiology to help with understanding the underlying problem.
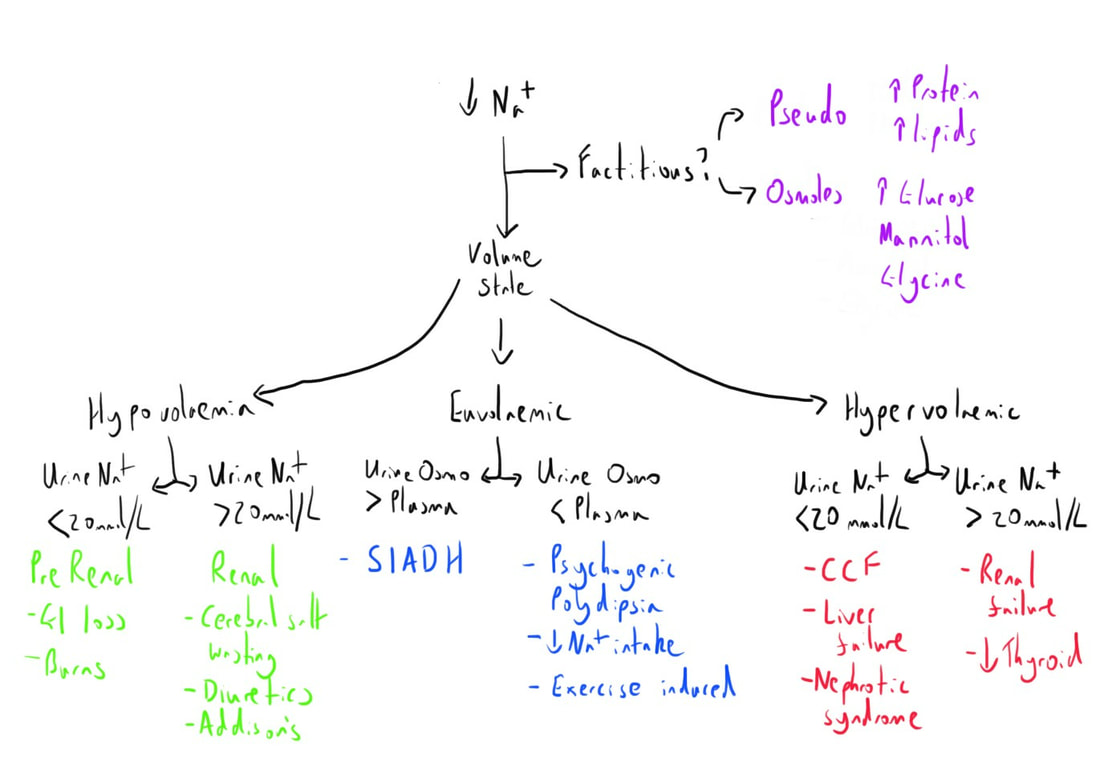
Management
This will be varied depending on the underlying cause and volume state.
A general rule is that the speed of correction should be proportional to the speed of disturbance.
Additional factors that may play a role include the degree of symptoms (often related to the speed of change), concomitant pathology e.g. intracranial, and the magnitude of the derangement.
The goals of treatment are:
Some general principles include:
Hypovolaemic
Treat with intravenous normal saline
Euvolaemic
Fluid restriction.
Demeclocycline may be used in SIADH.
Hypervolaemic
Treat underlying cause appropriately e.g. diuretic for CCF.
Chronic
Correct slowly, no more than 8 mmol/L over 24h. 4-6 mmol/L per 24h is advised.
The compensatory changes that have developed in the brain may result in pontine demyelinosis if correction is too quick.
Fluid restriction is commonly employed here.
Oral sodium supplementation may also be employed.
Acute (<48 hours)
Here the need for treatment is more urgent.
The development of cerebral oedema can lead to serious adverse outcomes.
Therefore correction to a ‘safe’ level is usually actively chased using hypertonic saline.
The severe manifestations can usually be improved with a rise of 4-6 mEq/L.
As such, there will tend to be a goal for rapid initial elevation, followed by allowing a period of stabilisation to avoid too rapid correction.
The other methods of allowing correction can then be employed once stability has been achieved.
Urgent treatment involves involves:
3% sodium can be safely given peripherally (UpToDate) and so delay to place a central line should not occur.
Repeated boluses may be needed if there is an ongoing failure for the sodium level to rise.
However, once an adequate daily rise has occurred, hypertonic saline can be discontinued.
A general rule is that the speed of correction should be proportional to the speed of disturbance.
Additional factors that may play a role include the degree of symptoms (often related to the speed of change), concomitant pathology e.g. intracranial, and the magnitude of the derangement.
The goals of treatment are:
- Prevent further decline in sodium
- Prevent brain herniation
- Relieve the symptoms
- Over overcorrection
Some general principles include:
- Correct underlying cause
- Treat acute severe hyponatraemia urgently
- Chronic hyponatraemia should be corrected slowly.
Hypovolaemic
Treat with intravenous normal saline
Euvolaemic
Fluid restriction.
Demeclocycline may be used in SIADH.
Hypervolaemic
Treat underlying cause appropriately e.g. diuretic for CCF.
Chronic
Correct slowly, no more than 8 mmol/L over 24h. 4-6 mmol/L per 24h is advised.
The compensatory changes that have developed in the brain may result in pontine demyelinosis if correction is too quick.
Fluid restriction is commonly employed here.
Oral sodium supplementation may also be employed.
Acute (<48 hours)
Here the need for treatment is more urgent.
The development of cerebral oedema can lead to serious adverse outcomes.
Therefore correction to a ‘safe’ level is usually actively chased using hypertonic saline.
The severe manifestations can usually be improved with a rise of 4-6 mEq/L.
As such, there will tend to be a goal for rapid initial elevation, followed by allowing a period of stabilisation to avoid too rapid correction.
The other methods of allowing correction can then be employed once stability has been achieved.
Urgent treatment involves involves:
- Critical care environment
- 150ml 3% sodium chloride IV over 20 minutes
- Assess sodium level
- Repeat 150ml 3% sodium chloride bolus
3% sodium can be safely given peripherally (UpToDate) and so delay to place a central line should not occur.
Repeated boluses may be needed if there is an ongoing failure for the sodium level to rise.
However, once an adequate daily rise has occurred, hypertonic saline can be discontinued.
Epidemiology
It is the most common electrolyte abnormality in clinical practice.
Its incidence is very difficult to assess due to this frequency and the subsequent underreporting.
Its incidence is very difficult to assess due to this frequency and the subsequent underreporting.
Complications
These include:
- Cerebral oedema
- Osmotic demyelination syndrome a.k.a. Central pontine demyelinosis
Cerebral Oedema
This can arise from the osmotic shifts that are associated with hyponatraemia.
With the reduced osmotic pressure now being exerted by the extracellular fluid (as sodium is the main osmotic substance) fluid moves into the brain tissue.
This can be particularly problematic in those patients in whom there is already brain swelling e.g. post trauma.
Preventing this and its complications, e.g. herniation syndromes, is an important goal of treatment.
It is more likely in acute hyponatraemia when there is little time for compensatory changes in osmotically active substances.
With the reduced osmotic pressure now being exerted by the extracellular fluid (as sodium is the main osmotic substance) fluid moves into the brain tissue.
This can be particularly problematic in those patients in whom there is already brain swelling e.g. post trauma.
Preventing this and its complications, e.g. herniation syndromes, is an important goal of treatment.
It is more likely in acute hyponatraemia when there is little time for compensatory changes in osmotically active substances.
Osmotic Demyelination Syndrome
This was previously described as central pontine myelinolysis.
It results from too rapid correction of hyponatraemia.
This is due to the fact that brain cells very rapidly adapt to the changed osmotic state imposed by hyponatraemia, primarily through loss of their own osmoles.
When the hyponatraemia is subsequently corrected, the cells are hypotonic compared to the extracellular fluid, and so they dehydrate, with demyelination being a feature of the result cell damage.
The name has been changed to represent that fact that this demyelination is not limited to the pons, but can be more diffuse.
The scenarios where rapid correction is more likely to occur include:
The clinical features are usually delayed by 2 to 6 days after the correction process and are often permanent in nature.
They include:
MRI scanning can identify features that are associated with the condition, although these may not become apparent until up to a month afterwards.
It results from too rapid correction of hyponatraemia.
This is due to the fact that brain cells very rapidly adapt to the changed osmotic state imposed by hyponatraemia, primarily through loss of their own osmoles.
When the hyponatraemia is subsequently corrected, the cells are hypotonic compared to the extracellular fluid, and so they dehydrate, with demyelination being a feature of the result cell damage.
The name has been changed to represent that fact that this demyelination is not limited to the pons, but can be more diffuse.
The scenarios where rapid correction is more likely to occur include:
- Hyponatraemia secondary to dehydration
- In adrenal insufficiency
- Very low sodium (<105 mEq/L)
- Malnourishment
- Hepatic disease (acute or chronic)
- Hypokalaemia
The clinical features are usually delayed by 2 to 6 days after the correction process and are often permanent in nature.
They include:
- Quadriparesis/paraparesis (including locked-in)
- Seizures
- Confusion
- Coma
- Dysarthria
- Dysphagia
- Movement disorders
- Behavioural disturbances
MRI scanning can identify features that are associated with the condition, although these may not become apparent until up to a month afterwards.
Specific Causes
A few important causes of hyponatraemia will be discussed in more detail here.
SIADH is a very important cause and discussed in notably more detail here: http://www.thegasmanhandbook.co.uk/siadh.html
SIADH is a very important cause and discussed in notably more detail here: http://www.thegasmanhandbook.co.uk/siadh.html
Factitious
This represents a group of patients in whom the hyponatraemia is not due to an excess of water in relation to the sodium level i.e. dilution.
This may be more accurately called hyponatraemia without hypotonicity, and includes pseudohyponatraemia when it is due to the interference of an abnormality on laboratory equipment.
In other cases, the hyponatraemia arises because of the presence of a significant amount of another osmotically active agent.
Here is may be more useful to use the term tonicity.
Whilst osmolality considers all osmotically active molecules, tonicity makes special differentiation relating to those which do not cross the cell membranes.
For molecules that this does not refer to e.g. urea, a change in the level will affect the osmolarity, but this change will be uniform across the body compartments, and thus not lead to water shift between the compartments (as urea can also enter the cells easily).
In contrast, sodium is purely extracellular, and with a strong osmotic effect, and so a drop in this level reduces the tonicity of the extracellular fluid (in comparison to the intracellular compartment).
This difference will lead to water movement by osmosis.
The point of this is that in this group of hyponatraemia, there is not a change in the tonicity of the extracellular fluid, as the reduction in sodium osmotic effects is being replaced by another osmotically active molecule.
Indeed it is the increase in this molecule that leads to water moving into the compartment, ‘diluting’ the sodium.
There are a number of molecules that may result in this:
This may be more accurately called hyponatraemia without hypotonicity, and includes pseudohyponatraemia when it is due to the interference of an abnormality on laboratory equipment.
In other cases, the hyponatraemia arises because of the presence of a significant amount of another osmotically active agent.
Here is may be more useful to use the term tonicity.
Whilst osmolality considers all osmotically active molecules, tonicity makes special differentiation relating to those which do not cross the cell membranes.
For molecules that this does not refer to e.g. urea, a change in the level will affect the osmolarity, but this change will be uniform across the body compartments, and thus not lead to water shift between the compartments (as urea can also enter the cells easily).
In contrast, sodium is purely extracellular, and with a strong osmotic effect, and so a drop in this level reduces the tonicity of the extracellular fluid (in comparison to the intracellular compartment).
This difference will lead to water movement by osmosis.
The point of this is that in this group of hyponatraemia, there is not a change in the tonicity of the extracellular fluid, as the reduction in sodium osmotic effects is being replaced by another osmotically active molecule.
Indeed it is the increase in this molecule that leads to water moving into the compartment, ‘diluting’ the sodium.
There are a number of molecules that may result in this:
- Glucose
- Mannitol
- Glycine
- IV immunoglobulins
Pseudohyponatraemia
Here there isn’t actually a reduction in the sodium concentration in the water, but when the plasma is measured as a whole, the large volume of another substance there makes it appear (to some analysers) that the sodium concentration is actually reduced.
This doesn’t happen in analysers that look only at the water component rather than the plasma as a whole.
Cases where this may occur include:
This doesn’t happen in analysers that look only at the water component rather than the plasma as a whole.
Cases where this may occur include:
- Hyperlipidaemia
- Obstructive jaundice
- Plasma cell dyscrasias
Hypovolaemic
Here the patient has lost water and electrolytes, but the total sodium loss from the body is in excess of the water loss.
This results in less sodium per unit volume of water and so hyponatraemia.
A useful categorisation is between renal and prerenal causes.
This results in less sodium per unit volume of water and so hyponatraemia.
A useful categorisation is between renal and prerenal causes.
Prerenal
In prerenal cases, the sodium and water loss has been through another route.
The kidneys here can be thought of as functioning normally, and so urinary electrolyte analysis should demonstrate that they are trying to hold on to sodium, and so the measured levels will be low (<20mmol/L).
Examples include:
The kidneys here can be thought of as functioning normally, and so urinary electrolyte analysis should demonstrate that they are trying to hold on to sodium, and so the measured levels will be low (<20mmol/L).
Examples include:
- Third space losses
- Burns
- Bowel obstruction
- Pancreatitis
- Burns
- GI losses
Renal
In renal causes, there is failure of sodium conservation at the level of the kidney.
This failure results in sodium loss, and a subsequent urinary water loss because of the osmotic effect.
This can be identified through the presence of a larger than expected amount of sodium in the urine (>20mmol/L) as if the kidneys were functioning physiologically they should be maximally retaining sodium as part of attempts to maintain homeostasis.
Examples include:
This failure results in sodium loss, and a subsequent urinary water loss because of the osmotic effect.
This can be identified through the presence of a larger than expected amount of sodium in the urine (>20mmol/L) as if the kidneys were functioning physiologically they should be maximally retaining sodium as part of attempts to maintain homeostasis.
Examples include:
- Addison’s disease
- Diuretic therapy
- Renal failure
- Salt losing nephropathy
- Cerebral salt wasting
Cerebral Salt Wasting
This is very similar to SIADH in almost all regards.
The key distinguishing feature is that the pathophysiology leads to a hypovolaemic state, unlike the euvolaemic state of SIADH.
It usually follows intracranial surgery.
It is usually self limiting an is just treated with normal saline infusion.
The key distinguishing feature is that the pathophysiology leads to a hypovolaemic state, unlike the euvolaemic state of SIADH.
It usually follows intracranial surgery.
It is usually self limiting an is just treated with normal saline infusion.
Euvolaemic
Here the volume status will be ‘normal’ meaning that sodium has been lost whilst through some method there has been relative preservation of water.
Testing the urine osmolality here can be useful, as comparison against the plasma provides insight into the process that are going on.
Testing the urine osmolality here can be useful, as comparison against the plasma provides insight into the process that are going on.
Urine osmolality > plasma
This means that the urine is continuing to reabsorb water from the filtrate, thus producing concentrated urine, even despite the hyponatraemic state.
It is important to note that this hyponatraemic state should be triggering the normal homeostatic mechanisms, and so this clinical picture suggest that this is not happening.
As the normal response would be a reduction in ADH secretion, this is SIADH.
It is important to note that this hyponatraemic state should be triggering the normal homeostatic mechanisms, and so this clinical picture suggest that this is not happening.
As the normal response would be a reduction in ADH secretion, this is SIADH.
Urine osmolality < plasma
Here the urine is trying to keep itself dilute, and thereby get rid of the excess water.
This suggests that the homeostatic mechanisms are functioning, and hence the preserved volume state, but that an ongoing physiological stress is still leading to hyponatraemia which is not being fully compensated for.
In essence, the body is receiving more water than it can adjust for, or it is receiving less sodium than it can compensate for.
Causes include:
Tea and toast diet/beer diet
This essentially refers to inadequate dietary sodium intake.
The name refers to common presentations that this may take.
Both ‘diets’ have very low sodium, whilst usually preserving water intake.
They both are also protein low, and thus also reducing the solute load for excretion.
Psychogenic polydipsia
This refers to an increased oral intake of fluid due to an abnormal thirst response.
This is commonly associated with psychiatric disorders.
In essence, the threshold for thirst is adjusted from the normal baseline, causing patients to still feel thirst at a lower plasma osmolality.
Normally the reduction in ADH secretion that would accompany the increased fluid intake could compensate and this result in only a minor sodium change, albeit with a notable polyuria.
There would appear to be some disturbance of this homeostatic process in those patients who develop hyponatraemia.
This probably relates to abnormal ADH response i.e. a form of SIADH.
Exercise induced hyponatraemia
This is a phenomenon seen is extreme sports participants.
Here the water intake is high in comparison to the sodium losses e.g through sweating.
Even electrolyte balanced drinks may be unable to fully keep up with these losses.
There may also be some SIADH in response to the exercise itself (e.g. triggered by pain) and thus impaired water excretion.
This suggests that the homeostatic mechanisms are functioning, and hence the preserved volume state, but that an ongoing physiological stress is still leading to hyponatraemia which is not being fully compensated for.
In essence, the body is receiving more water than it can adjust for, or it is receiving less sodium than it can compensate for.
Causes include:
- Psychogenic polydipsia
- Iatrogenic ‘water’ administration
- Inadequate dietary sodium - Tea and toast diet/beer diet
- Exercise induced hyponatraemia
Tea and toast diet/beer diet
This essentially refers to inadequate dietary sodium intake.
The name refers to common presentations that this may take.
Both ‘diets’ have very low sodium, whilst usually preserving water intake.
They both are also protein low, and thus also reducing the solute load for excretion.
Psychogenic polydipsia
This refers to an increased oral intake of fluid due to an abnormal thirst response.
This is commonly associated with psychiatric disorders.
In essence, the threshold for thirst is adjusted from the normal baseline, causing patients to still feel thirst at a lower plasma osmolality.
Normally the reduction in ADH secretion that would accompany the increased fluid intake could compensate and this result in only a minor sodium change, albeit with a notable polyuria.
There would appear to be some disturbance of this homeostatic process in those patients who develop hyponatraemia.
This probably relates to abnormal ADH response i.e. a form of SIADH.
Exercise induced hyponatraemia
This is a phenomenon seen is extreme sports participants.
Here the water intake is high in comparison to the sodium losses e.g through sweating.
Even electrolyte balanced drinks may be unable to fully keep up with these losses.
There may also be some SIADH in response to the exercise itself (e.g. triggered by pain) and thus impaired water excretion.
Hypervolaemia
Here there is an excess of total body water.
This excess water is essentially diluting the remaining sodium, leading to a hyponatraemia.
Again, a useful assessment can be based around how the kidneys are responding.
A low urinary sodium suggests sodium retention by the kidneys, whilst a high sodium suggests ongoing loss.
This excess water is essentially diluting the remaining sodium, leading to a hyponatraemia.
Again, a useful assessment can be based around how the kidneys are responding.
A low urinary sodium suggests sodium retention by the kidneys, whilst a high sodium suggests ongoing loss.
Low urinary sodium (<20 mmol/L)
This clinical picture suggests that the kidneys are retaining sodium.
This may be part of the pathology, in the sense that it is part of a now maladaptive response to a problem (e.g. inadequate intravascular pressures).
Causes include:
CCF/Liver failure
Here the pathophysiology is similar.
The starting point is an abnormality of the intravascular state.
In CCF this is due to the poor contractile function of the heart, and in liver failure it is due to vasodilation.
Low stimulation of the normal homeostatic mechanisms e.g. carotid baroreceptors, triggers fluid retention in order to improve the circulation.
The result is an overall retention of water that is primarily extravascular, and which dilutes the sodium.
This may be part of the pathology, in the sense that it is part of a now maladaptive response to a problem (e.g. inadequate intravascular pressures).
Causes include:
- CCF
- Liver failure
- Nephrotic syndrome
- Hepato-renal syndrome
- Low albumin
CCF/Liver failure
Here the pathophysiology is similar.
The starting point is an abnormality of the intravascular state.
In CCF this is due to the poor contractile function of the heart, and in liver failure it is due to vasodilation.
Low stimulation of the normal homeostatic mechanisms e.g. carotid baroreceptors, triggers fluid retention in order to improve the circulation.
The result is an overall retention of water that is primarily extravascular, and which dilutes the sodium.
High urinary sodium (>20 mmol/L)
Here there is ongoing sodium loss in the urine.
Causes include:
Causes include:
- Renal failure
- Hypothyroidism
- Early diuretics
Links & References
- Newson, L. Hyponatraemia. Patient.info. 2016. https://patient.info/doctor/hyponatraemia-pro
- CritIC. Hyponatremia - a practical approach. Youtube. 2017. https://www.youtube.com/watch?v=dmM2K50bnKw
- Cadogan, M. Hyponatreamia. LITFL. 2016. https://lifeinthefastlane.com/investigations/hyponatraemia/
- Heaton, T. Fluid & Electroyte Overview. MedicalPhysiology.co.uk. 2019. http://www.medicalphysiology.co.uk/fluid--electrolyte-overview.html
- Hirst, C. et al. The adult patient with hyponatraemia. 2015. CEACCP. 15(5):248-252. https://academic.oup.com/bjaed/article/15/5/248/240571
- Sterns, R. Diagnostic evaluation of adults with hyponatremia. UpToDate. 2017.
- Sterns, R. Causes of hypotonic hyponatremia in adults. UpToDate. 2018.
- Hoorn, E. Stern, R. Causes of hyponatraemia without hypotonicity (including pseudohyponatraemia). UpToDate. 2017.
- Sterns, R. Osmotic demyelination syndrome (ODS) and overly rapid correction of hyponatraemia. UpToDate. 2018
